Pneumologie
Publié le 19 nov 2020Lecture 7 min
Diagnostic et prise en charge de l’histiocytose langerhansienne pulmonaire de l’adulte
Amira BENATTIA*, Abdellatif TAZI*/** - *AP-HP, Centre de référence des histiocytoses, service de pneumologie, hôpital Saint-Louis, Paris **Université de Paris, INSERM U976, Institut de recherche Saint-Louis, Paris
L’histiocytose langerhansienne (HL) est une maladie rare, cosmopolite, d’expression clinique variable. L’atteinte pulmonaire (HLP) est le plus souvent isolée chez l’adulte et touche électivement les jeunes fumeurs. Le sevrage tabagique est essentiel et constitue la principale mesure initiale de la prise en charge associée à une surveillance rapprochée dans la plupart des cas. Pour les formes progressives avec altération de la fonction respiratoire, la cladribine constitue une avancée thérapeutique. Les progrès récents dans la connaissance de la pathogénie de l’HL ouvrent la voie aux traitements ciblés.
Qu'est-ce que l'histiocytose langerhansienne ?
L’HL est une maladie rare, d’étiologie indéterminée, caractérisée par une infiltration des organes atteints par des cellules dérivant des cellules dendritiques. Elle peut s’observer à tout âge et survient de façon presque toujours sporadique. En France, son incidence annuelle chez l’enfant de moins de 15 ans est de 4 à 5 cas/million et de 2 à 3 cas/million chez l’adulte. On distingue les formes mono-tissulaires, touchant un seul organe, le plus souvent l’os et le poumon chez l’adulte, des formes systémiques qui concernent au moins deux organes, notamment l’os, la peau, la tige pituitaire (diabète insipide) et le poumon. La maladie peut passer d’une forme localisée à une forme systémique. L’atteinte du foie, de la rate ou hématologique a un pronostic défavorable (organes dits à risque).
L’atteinte pulmonaire de l’HL peut s’inscrire dans le cadre d’une forme systémique, observée à tout âge, ou être isolée. C’est cette forme que l’on rencontre en milieu pneumologique et qui fait partie du spectre des pneumopathies infiltrantes diffuses (PID) kystiques. Elle survient quasi exclusivement chez des jeunes fumeurs ou ex-fumeurs des deux sexes, avec un pic de fréquence entre 20 et 40 ans. Une proportion significative de patients consomme en plus du cannabis.
Quand y penser ?
L’HLP est le plus souvent révélée par des symptômes respiratoires non spécifiques (toux, dyspnée d’effort, douleur thoracique, sifflements respiratoires). Des signes généraux, asthénie, amaigrissement, sueurs, fièvre sont associés aux signes respiratoires dans 15 à 20 % des cas. Elle peut aussi être découverte à l’occasion d’un pneumothorax spontané ou fortuitement à l’occasion d’une imagerie thoracique.
Quels outils pour le diagnostic ?
Un diagnostic présomptif d’HLP est souvent porté devant un tableau clinique compatible, associé à un aspect scanographique thoracique caractéristique. La nécessité d’une confirmation histologique se discute au cas par cas. La radiographie thoracique montre typiquement un syndrome réticulomicronodulaire symétrique prédominant dans les parties supérieures et moyennes des poumons.
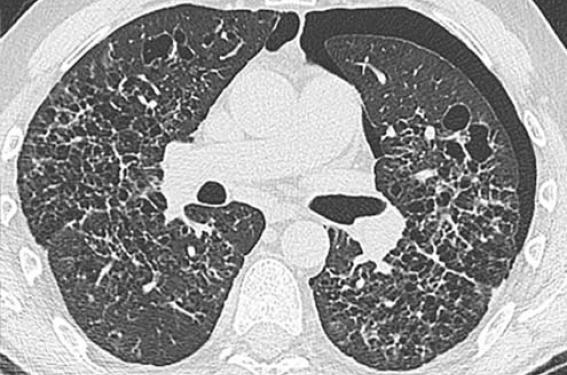
La tomodensitométrie (TDM) thoracique de haute résolution (TDMHR) est l’examen clé du diagnostic. Elle montre l’association de nodules dont certains sont cavitaires et de kystes à paroi épaisse ou fine, de taille variable, parfois confluents. Les lésions prédominent dans les parties supérieures et moyennes des poumons. Globalement, on distingue deux aspects : nodulo-kystique (figure 1) et kystique diffus (figure 2). On peut aussi observer un pneumothorax (figure 3) ou une lyse costale (figure 4).
Figure 1. TDM-HR thoracique d’une HLP récente, montrant un aspect nodulo-kystique typique.
Figure 2. TDM-HR thoracique d’une HLP ancienne avec un aspect purement kystique.
Figure 3. TDM-HR thoracique d’une HLP nodulokystique compliquée d’un pneumothorax gauche.
Figure 4. TDM-HR thoracique d’un patient ayant une PID nodulo-kystique. (A) Présence d’une lyse costale gauche ; mieux visualisée en fenêtre médiastinale (B).
La fibroscopie bronchique est macroscopiquement normale. Les biopsies de la muqueuse bronchique ne sont pas contributives pour le diagnostic. Le lavage broncho-alvéolaire (LBA) montre une alvéolite macrophagique, avec parfois une augmentation modérée des polynucléaires éosinophiles. La recherche de cellules CD1a+ est peu rentable.
La confirmation histopathologique est en règle obtenue par biopsie pulmonaire chirurgicale, parfois sur des biopsies transbronchiques. Les lésions sont caractérisées par l’accumulation de cellules qui ont l’aspect de cellules de Langerhans (d’où le terme d’HL) organisées en granulomes dans la paroi des bronchioles distales qu’elles détruisent. Ces cellules expriment le CD1a et la langerine (CD207) (figure 5). Les kystes sont le résultat de la destruction et de la dilatation des bronchioles.
Figure 5. Aspects histopathologiques des lésions pulmonaires de l’HLP. (A) Hématoxyline et éosine : lésion récente montrant des plages de cellules de grande taille à noyau plicaturé et à cytoplasme abondant clair. Il s’y associe de nombreux polynucléaires éosinophiles. (B) L’étude immunohistochimique montre que ces cellules de grande taille expriment le CD1a. Agrandissement x 200. (Images Dr Véronique Meignin)
La confirmation de l’HLP peut être obtenue par la biopsie d’un autre organe, à condition que le tableau pulmonaire soit compatible.
Quels sont les diagnostics différentiels ?
Devant un aspect nodulo-kystique à la TDM-HR thoracique, il faut éliminer une infection (mycobactéries, pneumocystose…), des métastases cavitaires, des emboles septiques, une sarcoïdose cavitaire ou une granulomatose avec polyangéite. La forme kystique diffuse peut être difficile à distinguer d’un emphysème ou d’une dilatation des bronches kystiques. On discute la lymphangioléimyomatose (LAM) chez la femme, le syndrome de Birt-Hogg-Dubé (BHD), la maladie de dépôts de chaînes légères non amyloïdes ou une pneumopathie interstitielle lymphoïde.
Quel bilan réaliser ?
Au diagnostic, un examen clinique complet est essentiel. Le bilan biologique standard peut montrer une discrète hyperleucocytose à polynucléaires neutrophiles ou un syndrome inflammatoire modéré. Le bilan hépatique peut objectiver une cholestase, qui doit faire rechercher une localisation hépatique spécifique.
L’exploration fonctionnelle respiratoire (EFR) montre habituellement une capacité vitale diminuée, un volume résiduel normal ou augmenté, une capacité pulmonaire totale normale et un rapport VR/CPT normal ou augmenté (piégeage). Un trouble ventilatoire obstructif et plus rarement un trouble ventilatoire restrictif peuvent être observés. La DLCO est abaissée dans la majorité des cas. Les gaz du sang, le test de marche de six minutes et l’échographie cardiaque avec Doppler sont réalisés sur point d’appel (dyspnée importante, saturation pulsée en oxygène abaissée, atteinte sévère de la fonction respiratoire, atteinte scanographique étendue ou signes évocateurs d’une hypertension pulmonaire [HTP]).
Des investigations orientées sont réalisées en fonction des atteintes extra-thoraciques : radiographies osseuses avec panoramique dentaire, osmolarité sanguine et urinaire, IRM cérébrale et dosages hormonaux. Dans les formes systémiques, la 18F-FDG-TEP-TDM est utile pour le bilan d’extension.
Certains examens sont utiles en cas de diagnostic d’HLP incertain pour rechercher des pathologies alternatives : dosage sérique du vascular endothelial growth factor-D (LAM), étude du gène de la folliculine (BHD), électrophorèse des protéines plasmatiques et dosage de chaînes légères (PID kystique par dépôt de chaînes légères). Pour le suivi, l’examen clinique, la radiographie thoracique et l’EFR suffisent dans la majorité des cas.
Que sait-on de l'évolution ?
L’évolution de l’HLP est très variable : rémission spontanée ou après sevrage tabagique, évolution par poussées, et rarement aggravation rapide conduisant à une insuffisance respiratoire chronique. Environ la moitié des patients dégradent leur fonction respiratoire à 5 ans, et un sousgroupe de patients voit son VEMS diminuer de façon importante dans les deux ans suivant le diagnostic. Une HTP peut survenir au cours du suivi d’une HL ou être concomitante au diagnostic de l’HLP. La grossesse ne semble pas modifier le cours évolutif de l’HLP.
Dans les formes kystiques diffuses, le risque de pneumothorax au cours du travail doit être évalué.
Quelle prise en charge pour 2020 ?
Le sevrage tabagique constitue la pierre angulaire de la prise en charge. L’association de corticoïdes inhalés et de bêta-2- sympathomimétiques de longue durée d’action est utilisée à but symptomatique, et permet parfois d’améliorer la fonction ventilatoire. Les patients doivent être vaccinés (grippe et pneumocoque). La réhabilitation respiratoire est proposée dans certains cas. Les corticoïdes oraux améliorent les signes généraux, mais n’ont pas fait la preuve de leur efficacité sur l’atteinte respiratoire. La vinblastine, traitement de référence des formes systémiques de l’HL, est peu efficace dans l’HLP.
Dans les formes progressives, la cladribine, molécule de chimiothérapie de la classe des analogues des purines, a constitué une avancée importante dans le traitement des formes progressives d’HLP avec altération fonctionnelle.
Le pneumothorax nécessite souvent de recourir à la chirurgie thoracique (biopsie pulmonaire dans le même temps pour asseoir le diagnostic). Dans la mesure du possible, il faut éviter la pleurectomie, car une transplantation pulmonaire peut être nécessaire ultérieurement.
En cas d’HTP sévère, les traitements spécifiques vasodilatateurs artériels pulmonaires peuvent être utilisés avec prudence dans des centres spécialisés.
Une insuffisance respiratoire très sévère ou une HTP majeure peuvent conduire à la transplantation pulmonaire. Des rechutes de la maladie sur le greffon ont été rapportées.
Progrès thérapeutiques : vers des thérapies ciblées ?
La mise en évidence d’une activation constante de la voie des mitogen activated protein kinases (MAPK) dans les lésions d’HL a constitué une avancée majeure dans la compréhension de la pathogénie de la maladie (figure 6). Cette activation est le plus souvent due à une mutation de l’oncogène BRAF (mutation BRAFV600E), mais aussi à d’autres altérations (mutations/délétions de MEK, délétions de BRAF, mutations de NRAS). Dans environ 15 % des cas, le mécanisme responsable n’est pas connu.
Figure 6. Schéma simplifié de la voie des MAPK. (A) Activation physiologique. (B) Activation spontanée de la voie par les anomalies moléculaires décrites dans l’HLP, notamment la mutation BRAFV600E. Ligand du récepteur à tyrosine kinase (RTK) ; des mutations de MEK1 (MAP2K1) et de NRAS ont aussi été identifiées.
Cette découverte a ouvert la voie à une approche thérapeutique plus ciblée, par inhibiteur de BRAF ou de MEK. Ces traitements, qui ont des effets indésirables potentiellement sévères, sont réservés aux formes réfractaires d’HL et leur indication doit être discutée au cours de la RCP nationale du centre de référence (http://www.histiocytoses.fr).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :




