Publié le 21 sep 2023Lecture 11 min
Sarcoïdose pulmonaire
Pierre GAZENGEL*,**, F. JENY*, H. NUNES*, *Hôpital Avicenne, APHP, Bobigny, **Hôpital Marie-Lannelongue, Le Plessis-Robinson
La sarcoïdose est une pathologie inflammatoire de cause(s) inconnue(s) définie par la formation de granulomes au sein de divers organes, dont les atteintes pulmonaires et ganglionnaires représentent les localisations les plus fréquentes. Près de la moitié des patients évoluent spontanément favorablement, l’autre moitié des patients peut développer une atteinte chronique invalidante, dont la localisation pulmonaire est responsable dans plus de 90 % des cas, nécessitant un traitement par corticoïdes et/ou immunosuppresseurs.
ÉPIDÉMIOLOGIE
La sarcoïdose présente une prévalence en France de près de 30 cas pour 100 000 habitants. Au moment du diagnostic, près de 70 % des patients ont entre 25 et 45 ans chez les deux sexes, un second pic d’incidence est présent chez les femmes de plus de 50 ans. Cette pathologie se déclare rarement avant 15 ans et après 70 ans.
Diagnostic
Le diagnostic repose sur 3 éléments comprenant(1) :
– une présentation clinique et radiographique compatible ;
– la mise en évidence de granulome épithélioïde gigantocellulaire sans nécrose caséeuse (sauf exception, encadré 1) ;
– l’élimination des diagnostics différentiels (encadré 2, liste non exhaustive).
Clinique
Les symptômes respiratoires non spécifiques sont la toux, la dyspnée et de façon moins fréquente les sibilants ou rarement l’hémoptysie. La douleur thoracique est possible en présence d’adénopathies médiastinales et hilaires. L’hippocratisme digital est très rare. L’auscultation pulmonaire est paradoxalement normale en comparaison avec les anomalies radiographiques.
L’atteinte pulmonaire peut être isolée, ou s’accompagner d’atteintes extra-pulmonaires dans 30 à 50 % des cas. Tous les organes peuvent être touchés, avec le plus fréquemment des atteintes ganglionnaires, cutanées et ophtalmiques (tableau 1).
Enfin, des symptômes non liés à une atteinte spécifique directe d’organes (syndrome parasarcoïdien) comme la fatigue (70 % des cas), les douleurs (notamment dans le cadre d’une neuropathie des petites fibres) et les troubles cognitifs sont fréquents et peuvent considérablement impacter la qualité de vie des patients.
Histologie
Le site de prélèvement est orienté par la clinique et l’imagerie (éventuellement guidé par le TEP au 18-FDG si aucune cible n’est évidente à l’examen clinico-biologique ou scannographique) ainsi que l’accessibilité, le risque de complication, et la rentabilité (encadré 3).
Figure 1. Muqueuse bronchique en fond d’œil et sténose bronchique.
Bilan minimal initial
Chaque patient doit avoir un bilan initial minimal (encadré 4) qui vise à éliminer certains diagnostics différentiels, éliminer des atteintes sévères d’organes, évaluer l’activité de la maladie et à but préthérapeutique en recherchant des contre-indications à certains traitements (obésité, diabète et corticoïdes, désir d’enfant ou insuffisance rénale et méthotrexate…).
ÉPREUVES FONCTIONNELLES RESPIRATOIRES
Les épreuves fonctionnelles sont le plus souvent normales, ou peuvent révéler un trouble ventilatoire obstructif, restrictif ou une altération de la diffusion du CO.
Une hyperréactivité bronchique est retrouvée chez 20 % des patients, fréquemment associée avec une atteinte endobronchique. Le trouble ventilatoire obstructif peut être le reflet d’une sténose bronchique par compression extrinsèque ou par atteinte endobronchique granulomateuse, il est plus fréquent chez les patients atteints de fibrose en présence de distorsions bronchiques ou bronchectasies par traction.
Une désaturation au test de marche de 6 minutes peut être révélatrice d’une hypertension pulmonaire, la distance parcourue a une valeur pronostique de la maladie (distance < 300 m en cas d’hypertension pulmonaire). L’hypoxémie est mise en évidence chez des patients atteints de fibrose pulmonaire et/ou d’hypertension pulmonaire.
Imagerie
Radiographie thoracique
Quatre stades radiographiques selon la classification de Scadding ont été décrits et sont associés au risque de chronicité (tableau 2).
Tomodensitométrie thoracique
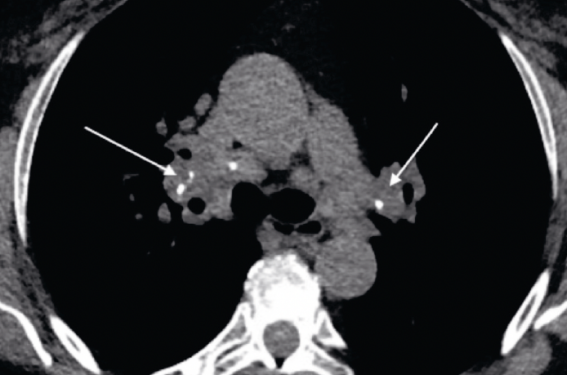
Les atteintes typiques de sarcoïdose pulmonaire sont(3) : Les adénopathies médiastinohilaires bilatérales, symétriques, plus ou moins calcifiées (forme chronique) (figure 2). Le caractère unilatéral persistant à 3 mois du diagnostic, ou l’asymétrie des adénopathies hilaires, ou des adénopathies médiastinales sans adénopathies hilaires doivent faire suspecter un diagnostic différentiel, de même que des adénopathies péricardiques ou mammaires internes.
Figure 2. Adénopathies médiastino-hilaires bilatérales calcifiées.
Le syndrome micronodulaire avec distribution lymphatique (péri-bronchovasculaire et sous-pleurale) avec un aspect perlé de la scissure (figure 3).
Figure 3. Scissure perlée.
Le signe de la galaxie est quasi pathognomonique : il s’agit d’un nodule entouré de multiples micronodules confluents (figure 4).
Figure 4. Signe de la galaxie.
Des lignes septales sont plus volontiers retrouvées aux apex. La fibrose pulmonaire liée à la sarcoïdose présente plusieurs patterns scannographiques :
– distorsion bronchique avec ou sans masses hilaires bilatérales avec engainement péri-bronchovasculaire, distorsion et sténoses bronchiques, veineuses et artérielles pulmonaires (figure 5) ;
– opacités linéaires : lignes septales distordues, lignes hilo-périphériques ;
– le rayon de miel à prédominance apicale, péri-bronchovasculaire et centrale, avec un aspect à large maille, contrairement à la pneumopathie interstitielle commune.
Figure 5. Masses de fibrose péri-hilaires.
TEP TDM
La tomographie par émission de positons (TEP-TDM) marquée au 18-F-fluorodéoxyglucose est un examen utile pour évaluer l’activité de la sarcoïdose, elle n’est pas nécessaire dans une majorité de cas, elle reste cependant indiquée dans certaines situations précises :
– recherche de cible extra-thoracique infraclinique en l’absence de preuve histologique ;
– évaluation de l’activité pulmonaire en cas de fibrose pulmonaire évoluée ;
– recherche d’atteinte cardiaque (régime spécifique +++) ;
– bilan d’une fatigue intense : lors d’une maladie semblant non évolutive avec des tests biologiques normaux pour préciser définitivement si une activité est encore présente ;
– en cas d’hypertension pulmonaire : recherche d’hypermétabolisme médiastinal d’adénopathies obstructives.
TRAITEMENTS
La question de la prise en charge thérapeutique des patients a fait l’objet de nouvelles recommandations par la European Respiratory Society(4) et la British Thoracic Society(5). Les premières reposent sur une méthodologie de type GRADE (Grading of Recommandations, Assessment, Development and Evaluation), et proposent qu’un traitement soit initié dont le but d’éviter une forme sévère et/ou améliorer la qualité de vie. Les secondes recommandations, plus pragmatiques, renseignent sur les doses et la durée des traitements.
L’effet de la corticothérapie et autres immunosuppresseurs/immunomodulateurs n’est pas curatif, mais uniquement suspensif de la réaction granulomateuse ; par ailleurs, il n’existe pas de données sur l’effet du traitement sur l’histoire naturelle de la maladie.
Le traitement n’est pas systématique et la décision de traiter nécessite de répondre à certaines questions au préalable :
– le patient est-il symptomatique et existe-t-il une altération de sa qualité de vie ?
– les symptômes peuvent-ils être contrôlés par une thérapeutique locale ?
– existe-t-il une atteinte sévère d’organe ?
Les patients à haut risque de sévérité pulmonaire sont ceux avec une altération de la fonction respiratoire se traduisant par une diminution de la CV et/ou de la DLCO (la BTS propose comme seuils une DLCO < 65 %, des volumes mobilisables < 70 % ou une réduction de 10 % de la CV ou de 15 % de DLCO) ; la présence d’hypertension pulmonaire, ou de fibrose pulmonaire modérée à sévère.
– le patient présente-t-il des contre-indications au traitement ?
– enfin, il est important de prendre en compte l’adhésion au traitement sur laquelle repose notamment une meilleure évolution de la qualité de vie sous traitement.
Corticoïdes
Le traitement de première intention reste la corticothérapie(4). La dose initiale préconisée est de 20 mg/j à 40 mg/jour pour les atteintes pulmonaires, suivie d’une décroissance progressive et un objectif à 10 mg/j dans les 3 mois. La durée de traitement est généralement d’au moins un an. En cas d’atteinte peu sévère, mais symptomatique, une plus faible de corticothérapie (10 mg/jour) peut être initiée.
D’autres molécules peuvent être prescrites(4), en cas de contre-indication, échec ou mauvaise tolérance des corticoïdes, d’invalidité permanente et maladie active malgré un traitement par corticoïdes seuls. Le traitement de seconde ligne après la corticothérapie est le méthotrexate en cas d’atteinte pulmonaire. Ce traitement présente moins d’effets secondaires infectieux que l’azathioprine. Il y a moins de données avec les autres immunosuppresseurs de 2e ligne. L’hydroxychloroquine peut être utilisée en première ligne avant la corticothérapie en cas d’atteinte cutanée modérée.
L’utilisation en première ligne d’un traitement immunosuppresseur comme le méthotrexate en remplacement de la corticothérapie (PREDMETH Study) ou de l’hydroxychloroquine en association avec la corticothérapie à visée d’épargne cortisonique (protocole QUIDOse) sont en cours d’essai. Les anti-TNF alpha sont un traitement de 3e intention. L’infliximab, l’adalimumab sont les deux molécules ayant fait preuve de leur efficacité, et l’infliximab le plus efficace. Ces traitements exposent à un haut risque infectieux et notamment de tuberculose. Une mise à jour du calendrier vaccinal et surtout un dépistage et un traitement d’une infection tuberculeuse latente est indispensable avant son instauration. On y associe une faible dose de méthotrexate ou aziathioprine ou corticoïdes pour limiter le risque d’effet paradoxal (granulomatose médicamenteuse) et limiter la survenue d’une résistance au traitement par le biais d’anticorps dirigés contre les anti-TNF. Tableau 3
D’autres molécules peuvent être utilisées en 4e ligne, ces molécules sont envisagées au cas par cas en cas d’échec ou d’intolérance des autres lignes thérapeutiques, et sur avis d’expert :
– Repository corticotropin injection : l’injection d’hormone adréno-corticotrope de l’hypophyse (ACTH) est proposée aux États-Unis, mais non disponible en France ;
– inhibiteurs de JAK ;
– rituximab
Réhabilitation respiratoire
Comme d’autres pathologies interstitielles pulmonaires fibrosantes, la réhabilitation respiratoire permet une diminution de la sensation de dyspnée, une prise en charge diététique, psychologique et des comorbidités du patient.
Transplantation pulmonaire
La sarcoïdose est à l’heure actuellement marginale dans les indications de transplantation pulmonaire. Elle peut cependant être proposée en cas de fibrose pulmonaire compliquée d’insuffisance respiratoire chronique terminale sous oxygénothérapie, et/ou d’hypertension pulmonaire. La survie en post-transplantation est comparable aux autres indications de transplantation pulmonaire, avec une médiane de survie à 8 ans(6).
FORMES PULMONAIRES SÉVÈRES
Fibrose pulmonaire
Près de 20 % des patients avec une sarcoïdose vont développer une fibrose pulmonaire qui peut dans certains cas entraîner une surmortalité, en raison de l’insuffisance respiratoire, de l’hypertension pulmonaire ou des complications infectieuses qui lui sont associées.
Les facteurs de risque de mortalité, notamment en cas de fibrose pulmonaire, sont une altération de la fonction respiratoire avec une élévation du composite physiologic index (CPI) > 40 (CPI = (91,0 -(0,65 x DLCO)- (0,53 x FVC) + (0,34 x FEV1)) ; une étendue supérieure à 20 % de la fibrose pulmonaire au scanner thoracique ou la présence d’hypertension pulmonaire, et l’origine afro-caribéenne.
Hypertension pulmonaire
L’hypertension pulmonaire associée à la sarcoïdose fait partie du groupe 5 de la classification. Les causes sont multiples et parfois intriquées : maladie parenchymateuse avec fibrose avancée, vasculopathie granulomateuse ; compression extrinsèque par les adénopathies médiastinales ou une médiastinite fibrosante ; cardiopathie sarcoïdosique ; comorbidités (SAOS, maladie thrombo-embolique) ; hypertension portale liée à une sarcoïdose hépatique. Une dyspnée, un test de marche de 6 minutes avec une distance parcourue < 450 m, une désaturation < 90 % en AA, un diamètre du tronc de l‘artère pulmonaire supérieur à celui l’aorte ascendante sur le TDM thoracique, une franche altération de la diffusion du CO (notamment contrastant avec une CV relativement conservée) doivent faire suspecter cette complication de la maladie, et conduire à réaliser une échocardiographie pour dépistage. L’examen de confirmation diagnostique reste le cathétérisme cardiaque droit. L’instauration d’un traitement spécifique de l’hypertension pulmonaire se discute auprès de centres experts(7). Les facteurs de surmortalité sont une DLCO < 35 % et/ou une distance parcourue au test de marche < 300 m.
Infections
Comme tout patient présentant une pathologie bronchique et pulmonaire fibrosante, plus ou moins associée à un traitement immunosuppresseur, les patients atteints de sarcoïdose pulmonaire sont sujets à des infections bactériennes (dont pyocyanique surtout en cas de bronchectasies) et virales respiratoires plus fréquentes et plus sévères que la population générale.
En dehors des stades I de la maladie, une mise à jour du calendrier vaccinal est requise (anti-Covid, grippale, pneumococcique).
Les aspergilloses pulmonaires chroniques (APC) sont volontiers rencontrées dans la sarcoïdose(8) et préférentiellement chez les patients présentant une fibrose pulmonaire avec des bronchectasies par traction et ceux exposés aux moisissures par leur profession. Dans une large série d’APC associées à la sarcoïdose, 77 % des cas étaient liés à Aspergillus fumigatus, 46 % des patients présentaient une co-infection. Les trois entités les plus retrouvées étaient l’aspergillose pulmonaire chronique cavitaire (APCC), l’aspergillose pulmonaire chronique fibrosante et l’aspergillome.
L’hémoptysie reste la principale complication, survenant dans la moitié des cas de façon concomitante d’une infection bactérienne. Une artério-embolisation bronchique est parfois nécessaire, le plus souvent en cas d’APCC. La chirurgie thoracique est exceptionnellement envisagée, le plus souvent à visée hémostatique en cas d’hémoptysie massive, il existe par ailleurs un haut risque de récidive d’APC au décours.
Les traitements azolés sont utilisés en première intention, et préférentiellement de voriconazole pour une durée de 6 à 12 mois. Un dosage de leur taux sérique est préconisé. La tolérance est imparfaite, avec la nécessité d’une interruption de traitement dans un tiers des cas, le plus souvent pour une toxicité hépatique ou cutanée. D’autres molécules comme le posaconazole et l’isavuconazole présentent un meilleur profil de tolérance et peuvent être également proposées. L’efficacité est évaluée par le taux sérique d’IgG anti-aspergillaire et l’épaisseur des parois des cavités aspergillisées.
PRONOSTIC
Le pronostic de la sarcoïdose est souvent favorable, notamment pour les patients atteints de sarcoïdose de stade I asymptomatique et de Löfgren. Le pronostic de la sarcoïdose est favorable dans 80 % des cas avec ou sans traitement. Dix à 20 % des patients vont garder des séquelles et 1-5 % des patients vont décéder de leur sarcoïdose. La survie des patients atteints de sarcoïdose ne nécessitant pas de traitement est similaire à celle de la population générale. Les causes de décès liées à la sarcoïdose sont d’origine pulmonaire liée à la fibrose pulmonaire et à l’hypertension pulmonaire dans les pays occidentaux, l’atteinte cardiaque est quant à elle la première cause de mortalité au Japon.
SUIVI
Tout patient présentant une sarcoïdose nécessite un suivi. La surveillance pneumologique s’effectue tous les 3 à 6 mois selon la gravité, l’évolutivité du patient. Une spirométrie et l’évaluation de la diffusion du CO sont préconisées, ainsi qu’une radiographie thoracique. Un scanner thoracique n’est pas nécessaire de façon systématique.
Un ECG annuel, associé à une recherche de signes fonctionnels cardiaques à l’interrogatoire doivent être systématiques.
L’ECA peut être informatif sur l’activité de la maladie pour certains patients. Un dosage de la calcémie corrigée, phosphatase alcaline, de la créatininémie doit être effectué annuellement(2).
Remerciements : Dr Marjorie Latrasse, radiologue
Les auteurs déclarent ne pas avoir de conflit d’intérêts en rapport avec cet article.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :




