Pneumologie
Publié le 18 jan 2021Lecture 10 min
Le spectre des pneumopathies interstitielles diffuses chroniques liées au tabac
Robert BARBIER*, Guillaume GRANIER**, Irina LATU*** - *Centre de pneumologie, Clinique Rhône Durance, Avignon ; **Anatomopathologie, Centre hospitalier, Avignon ; ***Pneumologie, Centre hospitalier, Avignon
Les pneumopathies interstitielles diffuses liées au tabac constituent un éventail complexe de pathologies qui reste à ce jour encore mal cerné. Les raisons d’une relative confusion sont multiples : un certain nombre de ces pneumopathies sont rares, la terminologie est volontiers empruntée aux anatomopathologistes et il a fallu en retrouver la correspondance radio-clinique, de nombreux acronymes en langue anglaise ont vu le jour et il a fallu créer des équivalents en français (tableau). En outre, il existe des chevauchements entre certaines de ces entités. Le diagnostic anatomopathologique formel nécessite de larges biopsies chirurgicales qui ne sont que rarement exécutées dans la pratique clinique et le diagnostic est donc souvent présomptif. Pour finir, de nouvelles entités sont décrites par les pathologistes comme le « Smoking Related Interstitial Fibrosis » sans que sa pertinence clinique ait été clairement reconnue.
Les différentes pneumopathies interstitielles diffuses chroniques liées au tabac
La dernière classification des pneumopathies interstitielles diffuses idiopathiques ATS/ERS date de 2013(1). Entre autres précisions par rapport à l’édition de 2002, elle introduit la notion de pneumopathies interstitielles idiopathiques liées au tabac (Smoking Related Idiopathic Interstitial Pneumonias ou SR-IIPs). Cette nouvelle classification (figure 1) restreint les pneumopathies interstitielles diffuses idiopathiques liées au tabac à la bronchiolite respiratoire avec pneumopathie interstitielle et à la pneumopathie interstitielle desquamative. L’histiocytose langheransienne, dont le lien épidémiologique avec un tabagisme n’est plus à démontrer, est classée avec les « autres formes de pneumopathie interstitielle diffuse » et la fibrose pulmonaire idiopathique souvent liée à un tabagisme est classée avec les pneumopathies interstitielles idiopathiques chroniques fibrosantes. Le syndrome fibrose emphysème n’a pas une place dédiée dans cette classification de même que le Smoking Related Interstitial Fibrosis (SRIF) ou fibrose interstitielle liée au tabac en français (FILT) dont la description est trop récente(2). Dans la littérature anglo-saxonne, ces différentes pneumopathies interstitielles chroniques liées au tabac sont regroupées sous le terme générique de Smoking Related Interstitial Lung Disease (SR-ILD), encore un acronyme qui ajoute à la confusion(2). Ce sont la bronchiolite respiratoire avec pneumopathie interstitielle, la pneumopathie interstitielle desquamative et l’histiocytose langerhansienne qui semblent les plus marquées par le tabagisme dans leur apparition et dans une certaine mesure dans leur résolution(2).
Figure 1. Classification ATS/ERS 2013
Identification des différentes pneumopathies interstitielles diffuses chroniques liées au tabac
L’histiocytose langerhansienne(2,3) est en règle assez facilement reconnaissable avec un profil tomodensitométrique et une évolution dans le temps caractéristiques(4) : micronodules centro-lobulaires à contours mal définis remplacés par (ou associés à) des nodules étoilés des champs pulmonaires supérieurs et moyens évoluant vers la cavitation puis souvent sous la forme de kystes qui confluent avec le temps en prenant des formes « bizarres ». Dans 90 % des cas, il s’agit de patients fumeurs assez jeunes (20-40 ans). Le diagnostic est volontiers radiologique, mais un LBA révélant plus de 5 % de cellules de Langherans (CD1a +) peut conforter le diagnostic (avec néanmoins une sensibilité assez faible). La biopsie pulmonaire est rarement nécessaire et se discute au cas par cas. Une HTAP est parfois rencontrée, conséquence d’une insuffisance respiratoire sévère observée chez une petite proportion de patients, mais aussi parfois d’une véritable vasculopathie. Une atteinte extrapulmonaire (diabète insipide, kystes osseux et lésions cutanées) complète le tableau chez près de 20 % des patients. La mutation de certains gènes comme celui du gène BRAF (mutation BRAF-V600E) chez près de 50 % des patients ouvre la porte à un espoir thérapeutique. Le sevrage tabagique est indiqué, mais il n’empêche pas toujours l’évolution péjorative de la maladie. Paradoxalement, la poursuite du tabagisme ne se traduit pas toujours par une aggravation de la maladie. Des poussées sont parfois décrites chez certains patients lors de la reprise du tabagisme.
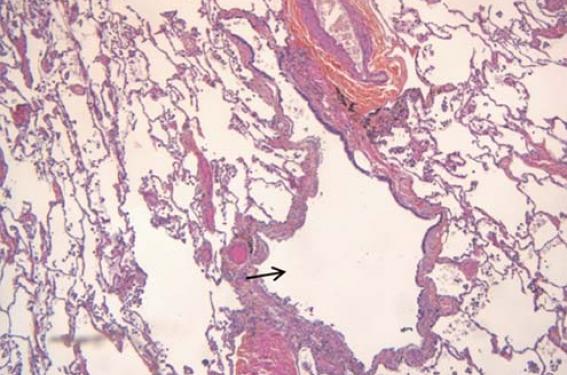
La bronchiolite respiratoire avec pneumopathie interstitielle (BRPI ou RB-ILD des Anglo-Saxons) touche presque exclusivement des gros fumeurs ou anciens fumeurs âgés de 30 à 60 ans(2,5,6). La bronchiolite respiratoire a une définition anatomopathologique. Elle se caractérise par le dépôt de macrophages pigmentés dans la lumière des bronchioles et des alvéoles adjacentes (figure 2). Le pigment est brunâtre et granuleux, on parle de « macrophages du fumeur ». La paroi des bronchioles est touchée par un processus inflammatoire modéré. Une discrète fibrose péribronchiolaire peut être observée(2,5). Le poumon à distance des bronchioles est épargné. Cette bronchiolite respiratoire est quasi constante chez les gros fumeurs et elle est souvent asymptomatique. Par convention(2), on parle de bronchiolite respiratoire avec pneumopathie interstitielle (BRPI) s’il y a un retentissement clinique (toux, dyspnée, râles crépitants des bases) ou fonctionnel (trouble ventilatoire obstructif et/ou restrictif). L’aspect en tomodensitométrie(4) dans la bronchiolite respiratoire avec ou sans pneumopathie interstitielle se caractérise par des images qui prédominent aux lobes supérieurs : nodules bronchiolocentrés volontiers flous, verre dépoli de répartition inégale avec parfois un aspect en mosaïque, épaississement de la paroi bronchique. Un emphysème peu marqué centrolobulaire ou paraseptal est fréquent. Le LBA témoigne d’une alvéolite à macrophages pigmentés. Une proportion importante de lym phocytes dans le LBA doit faire rechercher un diagnostic alternatif comme celui de pneumopathie d’hypersensibilité. La biopsie pulmonaire n’est pas né cessaire en dehors d’un doute diagnostic. Le sevrage tabagique s’accompagne d’une amélioration radio-clinique inconstante et qui peut être différée. Le pronostic reste néanmoins très favorable, il n’y a pas dans la littérature de description de formes fatales(2). La place des corticostéroïdes chez les rares patients qui s’aggravent n’est pas clairement définie. De même que la place de la biopsie pulmonaire chez ces mêmes patients.
Figure 2. Bronchiolite respiratoire : coloration hématoxyline-éosine, x 5, distension d’une bronchiole (flèche) avec présence dans la paroi de macrophages pigmentés et de lymphocytes (crédit : Guillaume Granier).
La pneumopathie interstitielle desquamative (PID ou DIP des Anglo-Saxons) offre les mêmes anomalies anatomopathologi - ques que la bronchiolite respiratoire, c’est-à-dire l’accumulation de macrophages pigmentés dans les bronchioles et les alvéoles(2,5-7). Mais ici l’accumulation est étendue et uniforme (non bronchiolo-centrée) avec inflammation des septa alvéolaires (figure 3). Un certain degré de fibrose est parfois visible que les anatomopathologistes néanmoins différencient bien de celle de la FPI. Le terme « desquamative » est impropre et tient au fait que les premières descriptions de Liebow évoquaient la desquamation de cellules épithéliales(2). Cette affection reste assez rare. Cliniquement, il s’agit de patients fumeurs (60 à 90 %) entre 40 et 60 ans, symptomatiques (dyspnée et toux d’apparition progressive). D’autres étiologies que l’exposition tabagique ont été identifiées (médicaments, exposition professionnelle, connectivite, infection…). Des râles crépitants sont souvent perçus. Il n’est pas rare de constater un hippocratisme digital. La fonction respiratoire est perturbée avec une amputation des volumes (modérée, voire absente), mais surtout avec une diffusion du CO très altérée. Une hypoxémie de repos est possible. Les images de tomodensitométrie(2,4-7) se caractérisent par des opacités en verre dépoli diffuses, parfois avec un aspect en mosaïque, à prédominance souspleurale et basale. Des petites images kystiques au sein du verre dépoli sont fréquentes de même qu’un emphysème paraseptal et centrolobulaire des lobes supérieurs. La présence de kystes en rayon de miel ou de réticulations est inhabituelle en dehors des formes qui évoluent de façon péjorative(2,8). Comme dans la bronchiolite respiratoire, le LBA témoigne le plus souvent d’une alvéolite à macrophages pigmentés. Il faut aussi rester vigilant si la proportion de lymphocytes est importante. L’évolution se fait en l’absence de traitement vers la détérioration, mais parfois aussi vers une stabilisation voire une régression des lésions sans qu’il ait été établi clairement que cela soit lié au sevrage tabagique. La possibilité de diagnostic alternatif et le pronostic conduisent à réaliser une biopsie pulmonaire chi rurgicale le plus souvent. Le trai tement repose sur la corti- cothé rapie, mais d’autres voies thérapeutiques ont été tentées(2,7). L’évolution de la maladie vers une insuffisance respiratoire terminale (ce qui oppose DIP et RB-ILD) conduit à proposer une transplantation avec un risque de récurrence de la maladie sur le poumon greffé déjà décrit dans la littérature(7).
Figure 3. Pneumopathie desquamative : coloration hématoxylineéosine, x 20, accumulation de macrophages pigmentés (flèche) dans les alvéoles (crédit : Guillaume Granier).
La fibrose pulmonaire idiopathique (FPI), la plus fréquente des pneumopathies intersti-tielles idiopathiques, possède souvent une présentation caractéristique avec un profil tomodensitométrique volontiers reconnaissable (gradient apicocaudal d’images de fibrose souspleurales comportant des réticulations intra-lobulaires, des bronchiolectasies et des images de kystes en rayon de miel). Cet aspect n’est pas spécifique de la FPI et peut s’observer dans d’autres situations (PID liée à une polyarthrite rhumatoïde par exemple ou pneumopathies d’hypersensibilité évoluées). Le diagnostic qui a fait l’objet de recommandations(9) repose sur un profil radiologique compatible et l’absence d’autres causes de PID identifiable. Il est de ce fait rare d’avoir recours à la biopsie chirurgicale. Le tabagisme est un facteur de risque indépendant pour la FPI. La proportion de fumeur y est de 41 à 83 % chez des patients de plus de 60 ans en général(2). Le tabagisme est aussi un facteur de risque d’exacerbation aigüe de FPI. Le pronostic est médiocre avec une survie médiane bien inférieure à 5 ans.
Le syndrome fibrose emphysème (ou CPFE des Anglo-Saxons) est décrit pour la première fois en 2005 par Cottin et coll.(2,10). Il se caractérise par des images de fibrose des bases pulmonaires (kystes en rayon de miel, réticulations et bronchectasies) et emphysème des sommets (paraseptal et centrolobulaire). Cette affection touche les hommes essentiellement après 60 ans. La combinaison d’une fibrose et d’un emphysème confère un profil fonctionnel original avec une spirométrie proche de la normale qui contraste avec l’aspect radiologique alors que la diffusion du CO est très altérée. La biopsie pulmonaire n’est pas conseillée. Le pronostic est médiocre et la survie est souvent inférieure à celle observée dans la FPI surtout en raison de l’existence d’une HTAP fréquente et de l’occurrence de cancer bronchique. Il n’y a pas de traitement dédié.
La fibrose interstitielle liée au tabac (SRIF des Anglo-Saxons) est décrite pour la première fois par les anatomopathologistes en 2010 comme une lésion assez fréquente dans les poumons de fumeurs ou anciens fumeurs(2,5,6,11,12). Il s’agit de découverte volontiers fortuite chez des patients devant subir une résection pulmonaire pour diverses raisons. Il est décrit anatomiquement un épaississement uniforme des septa alvéolaires par un collagène pauci-cellulaire et dense storiforme ou cireux, hyalinisé éosinophilique(11,12). S’y associent une bronchiolite respiratoire et un emphysème. Le tout pré- domine aux lobes supérieurs (figure 4, tirée de la référence 12, avec l’accord de l’auteur). De fait, cette forme particulière de fibrose au contact de lésions d’emphysème ou de bronchiolite respiratoire a pu être désignée dans la littérature sous les termes(2,5) de « airspace enlargement fibrosis » ou de « respiratory bronchiolitis with fibrosis ». Il peut exister une inflammation péribronchiolaire, mais elle reste alors à peine significative. La distribution des lésions est limitée à l’interstitium sous-pleural et péribronchiolaire sans jamais prendre un caractère diffus. Il n’y a pas ou très peu de foyer fibroblastique. Paradoxalement, ces lésions ont peu de conséquences cliniques et il n’est que très rarement observé d’évolution clinique perceptible. En TDM(2,4), on observe des réticulations, des nodules et des opacités en verre dépoli. Des images kystiques au sein du verre dépoli sont visibles également. Il s’agit de kystes à paroi fine prédominant dans les parties supérieures des lobes et un peu éloignés de la plèvre, ce qui les distingue des kystes en rayon de miel de la FPI(5). Identifier en anatomopathologie la FILT malgré un intérêt clinique encore mal défini semble important afin de ne pas la confondre avec d’autres pneumopathies interstitielles fibrosantes comme la pneumopathie interstitielle commune et la pneumopathie interstitielle non spécifique dans sa forme fibrosante. En effet, certaines PINS ont pu être reclassées comme des FILT après relecture(11).
Figure 4. Fibrose interstitielle liée au tabac (FILT – SRIF) : coloration hématoxyline-éosine, x 40, aspect caractéristique de collagène épais (flèche), storiforme et hyalinisé occupant les septa alvéolaires (crédit : Anna-Luise A. Katzenstein).
Les difficultés du diagnostic des pneumopathies interstitielles chroniques liées au tabac sont la règle(2,8). Elles posent le plus de problèmes à l’anatomopathologiste puis au clinicien. Rappelons que la bronchiolite respiratoire et la SRIF sont d’observation courante chez les fumeurs. Par ailleurs, pneumopathie desquamative et bron chiolite respiratoire peuvent coexister chez un même patient, certains considérant qu’il s’agit de formes extrêmes de la même entité(2). Ces mêmes lésions sont parfois observées chez les patients souffrant d’une HPCL typique(2). L’association de lésions de pneumopathie desquamative et de PINS chez un même patient est fréquente également(8). Enfin, il faudra à l’anatomopathologiste toute son attention pour ne pas confondre SRIF avec des lésions de FPI ou de PINS idiopathique fibrosante. De la même façon, les images tomodensitométriques sont rarement caricaturales et modérément spécifiques, des associations de pattern différents chez un même patient ne sont pas rares. C’est parfois l’évolution dans le temps des images qui permet de parvenir au diagnostic.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :




