Pneumologie
Publié le 01 déc 2017Lecture 9 min
La fibrose pulmonaire idiopathique : quand faut-il y penser ?
Mallorie KERJOUAN(a), Alexandre SALÉ(a), Stéphane JOUNEAU(a,b) a. Service de pneumologie, Centre de compétences pour les Maladies pulmonaires rares, Hôpital Pontchaillou, Rennes. b. IRSET UMR 1085, Université de Rennes 1, Rennes.
La fibrose pulmonaire idiopathique (FPI) est une maladie chronique d’évolution imprévisible au pronostic dramatique. Sa physiopathologie n’est pas encore totalement élucidée, toutefois le tabac joue probablement un rôle dans les agressions infracliniques répétées participant à la genèse de la maladie, tout comme les infections virales et bactériennes et la pollution. La présentation scanographique typique est celle d’une pneumopathie interstitielle commune (PIC) tout comme la présentation histologique typique. La médiane de survie serait de 3 à 5 ans, mais ces chiffres datent d’avant le développement des thérapeutiques antifibrosantes spécifiques. Chez les patients atteints de FPI légères à modérées, deux traitements antifibrosants ont montré une efficacité : la pirfénidone et le nintedanib. Ils ralentissent tous les deux le déclin de la fonction respiratoire. Le seul traitement curatif est la transplantation pulmonaire mais il ne s’adresse qu’à une minorité de patients très sélectionnés. Les complications de la FPI sont nombreuses, nécessitant un suivi et une prise en charge spécialisée.
Epidémiologie de la FPI
La fibrose pulmonaire idiopathique (FPI) est la forme la plus fréquente de pneumopathie infiltrante diffuse (PID) idiopathique chronique chez l’adulte(1). Les PID idiopathiques sont nombreuses et leur classification a été mise à jour en 2013(2). La FPI est une maladie rare. En France, la prévalence serait de 9 000 cas et l’incidence de l’ordre de 4 400 nouveaux cas par an. La FPI toucherait plus les hommes que les femmes (1,5 à 1,7/1) et augmente avec l’âge, la FPI étant rare avant 60 ans(3). La physiopathologie de la FPI n’est pas clairement élucidée mais les principaux facteurs de risque sont la fumée de cigarettes et les poussières de bois ou de métal(4). De même, les infections bactériennes et virales et la pollution jouent probablement un rôle dans les micro - agressions répétées participant à la genèse de la maladie. Au niveau cellulaire, l’apoptose des pneumocytes de type II et la prolifération des fibroblastes et myofibroblastes participent à la fibrogenèse(5) . La FPI est la forme la plus fréquente de pneumopathie infiltrante diffuse (PID) idiopathique chronique chez l’adulte et son incidence serait de l’ordre de 4 400 nouveaux cas par an en France.
Diagnostic de la FPI
Les patients atteints de FPI pré- sentent généralement une dyspnée d’effort qui s’aggrave progressivement ou une toux sèche, mais ils peuvent aussi être asymptomatiques. L’examen clinique révèle dans près de 100 % des cas des crépitants secs « velcro » prédominants aux bases à l’auscultation pulmonaire(6).
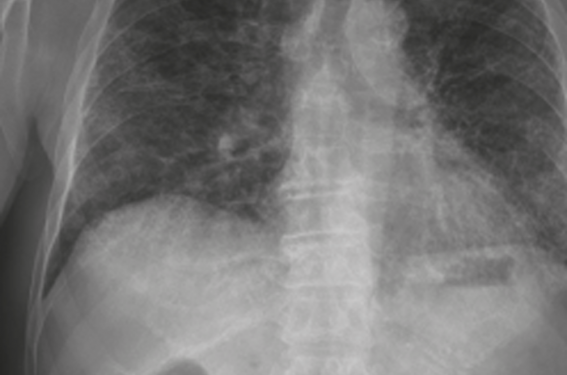
L’hippocratisme digital est moins fréquent, de l’ordre de 50 %(1). Le diagnostic est souvent tardif, car la dyspnée et les crépitants des bases chez un patient âgé font souvent penser à l’insuffisance cardiaque, certes beaucoup plus fréquente, mais, une fois ce diagnostic écarté, la possibilité d’une FPI n’est que rarement et trop tardivement évoquée. La présence de crépitants secs doit donc faire réaliser des investigations pneumologiques dès qu’une cause cardiaque est éliminée et faire évoquer une PID dès les consultations de soins primaires. La radiographie thoracique montre classiquement un syndrome interstitiel prédominant aux bases et en sous-pleural (figure 1) mais elle peut être mise en défaut dans les formes débutantes. Le scanner thoracique joue un rôle diagnostique majeur, les coupes fines étant indispensables pour une analyse précise. En effet, si les réticulations, à prédominance sous-pleurale et bi-basales, sont associées à la présence de rayon de miel et qu’il n’ y a pas d’atypies, il s’agit d’un tableau de pneumopathie interstitielle commune (PIC) certaine (figure 2, tableau), et l’obtention d’une histologie n’est pas nécessaire pour retenir le diagnostic de FPI (après avoir éliminé les diagnostics différentiels : médicaments pneumo toxiques (www.pneumotox.com), connectivites ou vascularites, expositions professionnelles ou domestiques)(1,4). Lorsque le scanner montre uniquement des réticulations sous-pleurales et bi-basales sans rayon de miel et sans atypie, on ne retient qu’un tableau radiologique de PIC possible (figure 3).
Pour prouver la FPI, il est alors nécessaire soit de réaliser une biopsie pulmonaire chirurgicale, soit de surveiller les patients avec des scanners thoraciques pour voir si le rayon de miel apparaît, mais cela ne concernerait que 53 % des patients à 6 ans, cette attitude pouvant donc entraîner un retard à la mise sous traitement spécifique adapté(7). En cas de signe(s) incompatible(s) avec le diagnostic radiologique de PIC, le diagnostic de FPI est le plus souvent écarté.
Les dossiers des patients doivent être analysés lors d’une discussion multidisciplinaire (DMD) incluant pneumologue(s), radiologue(s) et anatomopathologiste(s), idéalement spécialisés en pathologie thoracique, afin de la faire la synthèse clinique, fonctionnelle, biologique, radiologique voire histologique (figure 4) (1,4).
Cette étape est indispensable et doit être réalisée avant toute décision de biopsie pulmonaire chirurgicale et/ou avant toute mise en place de traitement antifibrosant spécifique. Si besoin, les dossiers pourront être présentés à une DMD de centre expert en PID. Les formes familiales et /ou génétiques de FPI représentent une minorité des cas (2 à 20 % en fonction des séries)(8). Il faut savoir y penser devant la présence d’un patient de moins de 50 ans, d’anomalies hématologiques, hépatiques et /ou cutanéomuqueuses. Les explorations fonctionnelles respiratoires (EFR) sont indispensables pour juger de la sévérité de l’atteinte et des critères d’éligibilité à un traitement antifibrosant spécifique. Le syndrome restrictif est la présentation fonctionnelle classique, associé à une altération des échanges gazeux. La capacité vitale forcée (CVF) et la capacité de transfert du monoxyde de carbone (DLCO) sont donc les deux éléments clés pour évaluer la gravité de l’atteinte. Une diminution précoce de la DLCO est souvent la seule anomalie détectée lors du diagnostic dans les formes précoces de FPI(1). Ces paramètres serviront également au suivi des patients. Associés à l’âge et au sexe, ils composent le score GAP (gender-age-physiology) qui a une valeur pronostique(9). Les crépitants secs « velcro » sont quasi constants chez les patients atteints de FPI. Le diagnostic peut être confirmé par le scanner thoracique lorsqu’il montre un aspect de PIC certaine, c’est-à-dire avec prédominance de réticulations aux deux bases et en sous-pleural avec rayon de miel et sans atypies. Le diagnostic TDM ainsi que la décision de réaliser une biopsie pulmonaire chirurgicale doivent être obligatoirement discutés lors d’une DMD (discussion multidisciplinaire).
Histoire naturelle de la FPI
La FPI est une maladie chronique, incurable (hors transplantation pulmonaire), s’aggravant progressivement de manière inéluctable mais d’évolution imprévisible (figure 5).
Certains patients vont en effet présenter un déclin progressif et lent de leur fonction respiratoire, alors que d’autres vont s’aggraver très rapidement. Des exacerbations aiguës (EA) de FPI sont également à craindre. Elles correspondent à une aggravation rapide de la dyspnée des patients, classiquement sur moins de 1 mois, avec l’apparition de nouvelles images bilatérales de type verre dépoli, soit sans cause retrouvée, soit dans un contexte infectieux ou postinfectieux, après avoir éliminé l’embolie pulmonaire (nécessité de réaliser un dosage de D-dimères et/ou un angioscanner thoracique) et l’insuffisance cardiaque (nécessité de doser les NT-proBNP et/ou réaliser une échocardiographie) (10) . Leur pronostic est excessivement sombre avec une médiane de survie de 3 à 4 mois. Des facteurs de risques d’EAFPI ont été identifiés : une CVF et une DLCO basses, une faible distance au test de marche de 6 minutes, une hypertension pulmonaire. Les EAFPI sont traitées par corticothérapie systémique à forte dose. Un essai thérapeutique est en cours (EXAFIP) testant l’ajout du cyclophosphamide contre placebo à la corticothérapie. Globalement, la médiane de survie des patients atteints de FPI serait de 3 ans après le diagnostic(5) . Cependant, cette notion mériterait d’être revue car date de l’étude des cohortes de patients qui n’avaient pas bénéficié des traitements antifibrosants spécifiques récemment mis sur le marché. L’évolution de la FPI est imprévisible. La médiane de survie des patients atteints de FPI serait de 3 ans après le diagnostic. Beaucoup de patients meurent des suites d’une exacerbation aiguë de FPI dont la médiane de survie est de 3 à 4 mois.
Pris en charge thérapeutique
La prise en charge thérapeutique comprend les vaccinations antigrippale et antipneumococcique, la réhabilitation respiratoire et les traitements symptomatiques (oxygénothérapie, antitussifs, etc.). La transplantation pulmonaire, seul traitement curatif, n’est possible que chez une minorité de patients, d’âge physiologique inférieur à 65 ans et sans comorbidité sévère. Il est ainsi recommandé d’adresser précocement ces patients dans un centre de transplantation pulmonaire pour avis. Les deux traitements antifibrosants ayant montré une efficacité sont la pirfénidone (ESBRIET® ) et le nintedanib (OFEV® ) (11-13). Ils permettent tous les deux de réduire approximativement de moitié le déclin de la fonction respiratoire (CVF). Les principaux effets indésirables de la pirfénidone sont cutanés (photosensibilité et éruption) et digestifs (anorexie, nausées, dyspepsie, diarrhée, cytolyse hépatique). Les principaux effets indésirables du nintedanib sont digestifs (diarrhée, nausées, vomissements, anorexie, amaigrissement, douleurs abdominales, cytolyse hépatique). Pour limiter les effets indésirables, les traitements doivent être pris au milieu des repas. Pour prescrire ces traitements antifibrosants, il faut que le diagnostic de FPI soit certain, que la CVF soit ≥ 50 %de la théorique et la DLCO ≥ 30 % de la théorique et qu’il n’y ait pas de contre-indications à leur utilisation. Des essais thérapeutiques ont testé la tolérance et la sécurité de l’association de ces deux molécules, mais les résultats sont en attente. Par ailleurs, des essais thérapeutiques sont en cours dans les formes débutantes (CVF ≥ 80 % théo) ou au contraire sévères (DLCO ≤ 35 % théo) avec les deux molécules antifibrosantes validées. Enfin, des essais thérapeutiques testant de nouvelles molécules sont en cours. Il est clairement recommandé de favoriser au maximum l’inclusion des patients dans toutes ces études afin d’améliorer la prise en charge thérapeutique des patients atteints de FPI. Les recommandations françaises proposent de traiter la FPI dès que le diagnostic est établi, en tenant compte de l’évaluation individuelle du bénéfice escompté et des risques du traitement(14). Deux traitements antifibrosants, la pirfénidone et le nintedanib, permettent de réduire de moitié le déclin de la fonction respiratoire. Il est recommandé de traiter les patients dès le diagnostic établi. Les inclusions dans les essais thérapeutiques sont à encourager. Un avis auprès d’un centre de transplantation est recommandé chez les patients d’âge physiologique < 65 ans sans comorbidités.
Complications de la FPI
De nombreuses complications peuvent apparaître dans le suivi des patients atteints de FPI. La toux sèche peut être invalidante et peut motiver une corticothérapie à faible dose en cas d’inefficacité de la codéine. L’hypertension pulmonaire peut être secondaire à la FPI, surtout en cas d’emphysème associé, mais doit faire éliminer une embolie pulmonaire, une insuffisance cardiaque gauche et un syndrome d’apnées obstructives du sommeil (SAOS). Le reflux gastro- œsophagien (RGO) est fréquemment associé à la FPI, et doit être le cas échéant traité. Le cancer bronchopulmonaire, fréquent dans cette population, pose des problèmes diagnostiques et thérapeutiques du fait de la FPI sous-jacente. En pratique, si l’on retrouve des crépitants secs chez un patient sans anomalie cardiaque, il faut éliminer une FPI. Le scanner thoracique est l’examen de choix qui devra être relu en DMD, si besoin dans un centre expert. La présence de réticulations aux deux bases pulmonaires n’est pas normale et doit faire évoquer une FPI débutante. Il faut savoir y penser si ces anomalies sont retrouvées sur les coupes parenchymateuses pulmonaires d’un scanner thoracique de dépistage du cancer bronchique ou d’un coroscanner ou de tout autre examen tomodensitométrique.
Conclusion
La FPI est une maladie incurable au pronostic sombre. L’évolution est imprévisible. Deux traitements antifibrosants sont disponibles, la pirfénidone et le nintedanib, qui permettent de ralentir de moitié le déclin de la fonction respiratoire. La recherche clinique continue et il faut inclure au maximum les patients dans les essais thé- rapeutiques afin de réussir à stabiliser et, peut-être un jour, guérir la FPI.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :